

Tirbanibulin是一种双重作用的 Src 激酶和微管蛋白聚合抑制剂,随着其相继在美国(2020年12月)和德国(2021年7月)批准上市用于治疗光化性角化病。微管蛋白双靶点抑制剂越来越受到科研人员的关注。
微管蛋白是癌症治疗的重要靶点,但微管靶向药物的耐药性和剂量限制性毒性限制了其临床疗效。近年来,多靶点治疗被认为是提高治疗效果的有效策略,特别是双靶点治疗。微管蛋白可与其它的具有协同效应的抗肿瘤药物联合治疗,因此设计双靶点微管蛋白抑制剂是克服耐药性、提高治疗效果的有效途径。
关于Tirbanibulin
1结构信息
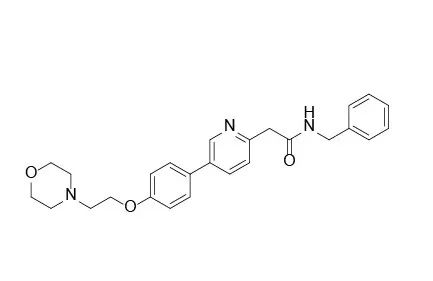
图1.Tirbanibulin结构式,来源:药渡数据
分子式:C26H29N3O3
分子量:431.53
CAS号:897016-82-9(KX 01)
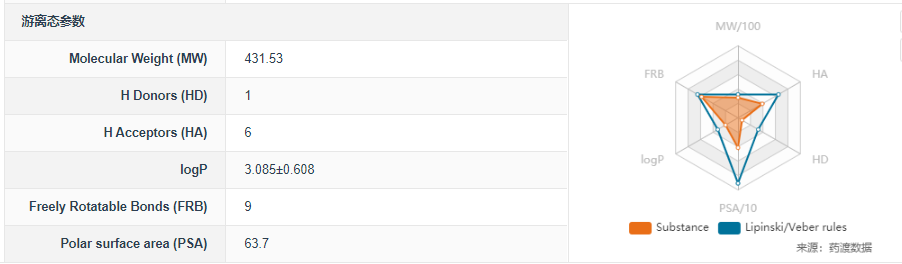
图2.Tirbanibulin离态参数,图片来源:药渡数据
2药物基本信息
Tirbanibulin是由Athenex Inc研发的是一种First-in-Class小分子药物,是一种SRC抑制剂和微管蛋白聚合抑制剂。目前该药物最高研发阶段为批准上市,用于治疗光化性角化病。
3上市信息
2020年12月14日,Tirbanibulin获得美国食品药品管理局FDA批准,由Athenex Inc销售,商品名为Klisyri®。(NDA213189)适应症为光化性角化病,为一种局部软膏剂,规格是1%。
2021年07月16日,Tirbanibulin获得欧洲药品管理局EMA批准,由Almirall Sa销售,商品名为Klisyri®。(EMEA/H/C/005183)适应症为光化性角化病,为一种油膏剂。
4研发里程碑
2017年09月15日,由Almirall Sa在美国开展临床三期试验,用于治疗光化性角化病。(NCT03285477;NCT03285490)
2016年04月11日,由Almirall Sa在美国开展临床二期试验,用于治疗光化性角化病。(NCT02838628)
2014年12月01日,由Athenex Inc在美国开展临床一期试验,用于治疗光化性角化病。(NCT02337205;NCT03575780)
2011年05月01日,治疗淋巴瘤和实体瘤的研究暂无进展。(NCT00658970)
2007年11月01日,由Athenex Inc在美国开展临床一期试验,用于治疗淋巴瘤和实体瘤。(NCT00658970)
双靶点微管蛋白抑制剂的提出
微管靶向剂(Microtubule-targeting agents,简称MTAs)能破坏微管的动力学和结构,进一步干扰有丝分裂纺锤体的形成,阻断有丝分裂中后期的细胞周期,诱导细胞凋亡。虽然在癌症治疗方面取得了巨大的成功,但其耐药性的产生阻碍了临床应用。肿瘤组织中β微管蛋白(尤其是βIII-tubulin)和膜结合药物外排蛋白如p糖蛋白(P-gp)的异常表达是MTAs的主要耐药机制,这些异常表达降低了肿瘤组织对MTAs的响应[1]。其他重要耐药机制包括微管与肌动蛋白相互作用的改变和凋亡通路的缺陷[2]。
影响多发性骨髓瘤临床疗效的另一个主要问题是其副作用,如神经系统和骨髓毒性的高发生率,继发性肿瘤的风险增加[3]。因此,研发新型MTA可以解决这些问题并提高患者存活率,是肿瘤治疗的一个新趋势。
双靶点抑制剂与单靶点药物相比克服了耐药性,通常可以改善治疗效果;与联合治疗中使用的多种药物相比,它们可能具有更容易预测的PK特征。此外,开发双靶点药物所需的临床试验可能少于联合治疗,而且其成本和风险与单靶点药物相似[4]。双靶点的选择通常是联合作用使表现出协同效应的靶点[5],常见的双靶点微管蛋白抑制剂会同时抑制微管蛋白和受体酪氨酸激酶(receptor tyrosine kinases inhibitor,RTK)、组蛋白去乙酰化酶(histone deacetylases inhibitor,HDAC)、雌激素受体(DNA-damaging agent,ER)或拓扑异构酶,具有优异的抗肿瘤活性[6]。
在双靶点药物协同作用的基础上,双靶点微管蛋白药物的开发引起了众多研究者的兴趣。由于双靶点药物的设计比单靶点药物的设计更为复杂,因此需要通过多种设计策略,包括药物再利用、双靶点抑制剂的骨架设计、基于药效团的联合和计算方法等,它们进一步加大了对双靶点药物的研发力度。
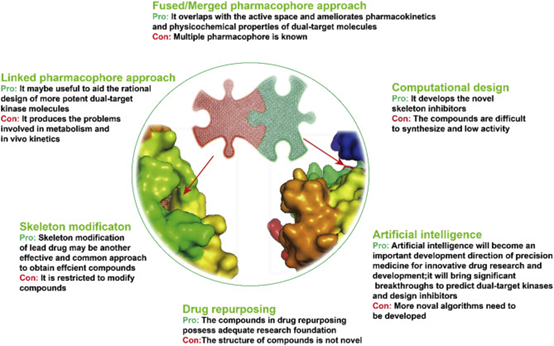
图3. 双靶点激酶药物设计方法的优缺点。图片来源:参考文献[4]
靶向微管的双靶抑制剂
1微管蛋白-RTK双靶点抑制剂
MTAs和RTKs(如VEGFR2、EGFR)抑制剂的联合治疗已在临床试验中显示出良好效果,包括头颈癌(NCT00720304和NCT00049283)、肝细胞癌(NCT00532441和NCT00553358)、 乳腺癌(NCT01050322和NCT00367471)和肺癌(NCT01405079)。因此,受联合治疗结果和双靶点药物策略优势的启发,研究人员设计确定了许多微管蛋白-RTK双重抑制剂。
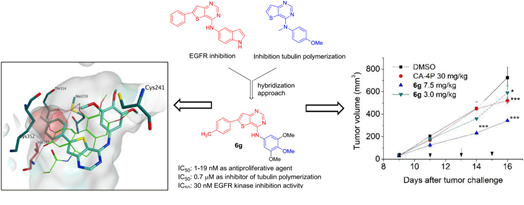
图4. EGFR微管蛋白双靶点抑制剂。图片来源:参考文献[7]
2019年,Romagnoli等人用微管蛋白抑制剂的3,4,5-三甲氧基苯胺基部分取代了VEGFR2-EGFR双重抑制剂C-4位的5-氨基吲哚侧链(图2所示),通过药效团合并的方法得到化合物6g,其与微管蛋白秋水仙碱位点结合并抑制微管蛋白组装,IC50值为0.71μM,EGFR(IC50 = 30 nM)抑制活性,但失去了对VEGFR2的效力。它在HeLa细胞中以剂量依赖性方式强烈抑制EGFR的磷酸化。化合物6g对多种癌细胞系具有强大的抗增殖活性(IC50 = 1-20 nM)。SAR研究表明,噻吩并[3,2-d]嘧啶支架对于抗增殖作用至关重要。在与噻吩并[3, 2-d]嘧啶核直接相连的苯环的对位引入CH3基团可以提高生物活性。用卤化物基团(Cl、Br和I)或OCH3 基团替换CH3基团也是可以容忍的,显示出有效的体外抗血管活性,与CA-4磷酸盐相比具有更有效的体内抗肿瘤作用,在7.5 mg/kg的剂量下具有52.5%的肿瘤生长抑制值。
2微管蛋白-HDAC双靶点抑制剂
HDACs可以调节染色质重塑和基因表达。靶向HDACs可间接调节癌症治疗中许多靶点的功能,包括微管中的α-微管蛋白、p53和Hsp90,这使得HDACs成为癌症治疗中的重要靶点。通常,HDAC抑制剂由锌结合基团(zinc-binding group,ZBG)、适当的接头和封端基团组成。HDAC抑制剂可以耐受各种封端基团,因此它们经常与其他癌症相关的靶标抑制剂杂交,用于开发双靶点药物。许多微管蛋白-HDAC双靶点抑制剂被开发出来以克服耐药性并提高抗肿瘤作用。
近期,Liou等人以ABT751(口服微管蛋白聚合抑制剂)的磺酰胺部分为封端基团,苯甲酰胺为ZBG,生成了一系列微管蛋白-HDAC双靶点抑制剂,如图3。其中,化合物19显著抑制KB癌细胞的生长(IC50 = 23 nM),甲氧基的去除或易位或置换降低了生物活性。将氮原子引入接头中产生化合物20,其显示出改善的抗增殖活性。
此外,化合物20有效抑制HDAC活性并在0.05μM时诱导微管组装的变化,在G2/M期触发细胞周期停滞并破坏微管动力学。将1-(芳基磺酰基)二氢吲哚骨架与苯甲酰胺基团融合,得到化合物21(图3)。化合物21可以显著抑制微管蛋白聚合(IC50 = 1.1 μM),并对HDAC1、-2和-6有抑制作用,IC50值分别为0.221、0.662和0.314 μM。此外,化合物21上调A549细胞中的乙酰化α-微管蛋白;对MDR阳性细胞系显示出有效的抗增殖活性,IC50值分别为64 nM (KB-VIN10)、43 nM (KB-S15)和46 nM (KB-7D);在A549和BJAB肿瘤异种移植模型中显示出有效的肿瘤抑制活性。
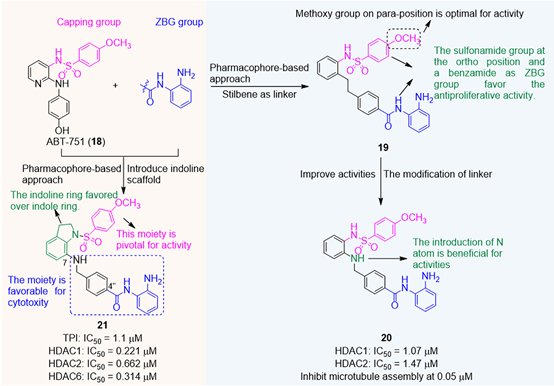
图5. 微管蛋白-HDAC双靶点抑制剂的设计,图片来源:参考文献[6]
3微管蛋白-雌激素受体双靶点抑制剂
雌激素受体(ER)是核受体超家族的一个分支,在大约75%的乳腺肿瘤中过表达,因此ER抑制剂可用于ER阳性的乳腺肿瘤治疗。紫杉醇与ER抑制剂Tamoxifen联合应用可提高其抗肿瘤作用。因此,开发微管蛋白-ER双靶点抑制剂是一种很有前途的抗肿瘤策略。基于结构的药物再利用策略被用来发现紫杉烷结合位点和ER之间的口袋相似性(图4A)。通过计算对接预测了几种潜在的选择性ER调节剂作为紫杉烷位点配体。体外试验表明,雷洛昔芬(RAL)可以增强微管稳定性,这与MSA紫杉醇相似。此外,基于细胞的图像分析表明RAL能够与SiR-微管蛋白竞争,SiR-微管蛋白是一种荧光紫杉醇偶联物,可能直接与紫杉烷位点结合。总的来说,RAL可以同时靶向ER和微管蛋白中的紫杉烷位点,用于治疗对紫杉烷位点突变具有抗性的癌症。
将三甲氧基芳基与配体中具有两个酚羟基的ER药效团结合,研发了具有β-内酰胺支架的双重微管蛋白-ER抑制剂(图4B)。最初,化合物53虽然表现出有效的ER 结合亲和力,但在50 μM时对MCF-7没有抗增殖活性。为了提高抗增殖活性并保持ER结合亲和力,制备了在β-内酰胺核心的C-3位具有α-(羟基芳基)甲基取代基的化合物54。它显示出优异的 ER结合亲和力,并提高了对MCF-7细胞的抗增殖活性(IC50 = 0.21 μM)。此外,化合物54是微管蛋白去稳定剂,导致微管蛋白解聚,细胞周期G2/M期停滞。
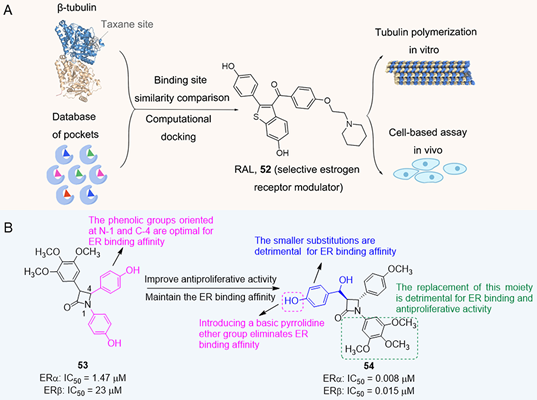
图6.微管蛋白-ER双靶点抑制剂,图片来源:参考文献[6]
微管蛋白-c-MET双靶点抑制剂
在肿瘤细胞中,肝细胞生长因子受体c-MET因发生突变或过度表达而表现出异常活化。Tivantinib是一种非ATP竞争性的c-MET抑制剂,其抗肿瘤活性已在临床试验中得到验证。研究发现Tivantinib有显著的抗增殖活性,并在G2/M期诱导细胞周期停滞,进一步分析发现Tivantinib可以破坏微管网络并抑制微管蛋白聚合。此外,Tivantinib可以规避ABC转运蛋白介导的多药耐药性。Tivantinib与微管蛋白复合物的X射线晶体结构显示Tivantinib可以与β-微管蛋白中的秋水仙碱位点结合,并与βN256 和βA315 的残基形成H键相互作用(PDB 5CB4,图5)。与c-MET激酶结构域复合的Tivantinib的晶体结构显示Tivantinib与一个新的c-MET口袋结合。该复合物与Met1160和Pro1158形成两个典型的H键铰链相互作用,并与Lys1161(PDB 3RHK)形成水介导的H键相互作用。目前,Tivantinib正处于单药治疗或与其他化疗联合治疗多种癌症的 II/III 期临床试验(NCT01755767、NCT01395758、NCT01447914和NCT01075048)。
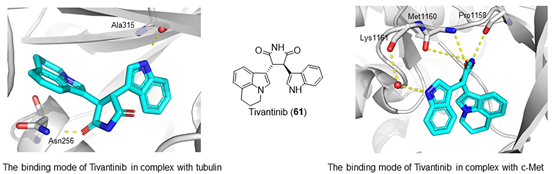
图7.微管蛋白-ER双靶点抑制剂,图片来源:参考文献[6]
微管蛋白-Katanin双靶点抑制剂
Katanin属于微管切断蛋白之一,在微管结构和方向的动态调节中发挥重要作用,以维持细胞稳态。破坏katanin的活性可影响微管结构,导致细胞凋亡,克服MTAs的耐药限制。基于组合原理,通过融合β-内酰胺型微管蛋白聚合抑制剂1和嘌呤型katanin激活剂2的结构,鉴定了具有咪唑并[4,5-c]吡啶-2-one支架的化合物20b可同时靶向微管蛋白和Katanin(图6)。化合物20b抑制微管蛋白聚合,IC50值为8.4μM,并具有中等结合亲和力,Kd值为12.7μM,显示出比化合物2更高的效力。此外,化合物20b具有良好的药代动力学特性(Cmax = 0.58μg/mL,t1/2 = 9.36 h,AUC0-24h = 2.63 mg·h/mL),可以抑制体内肿瘤生长,表明同时抑制微管蛋白和微管相关蛋白是一种有效的癌症治疗方法。
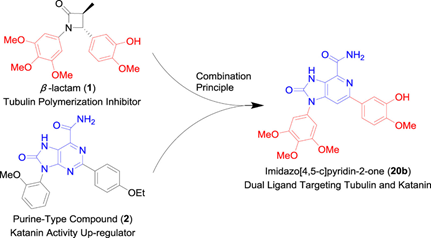
图8.微管蛋白-Katanin双靶点抑制剂,图片来源:参考文献[8]
近年来,调控多种肿瘤相关靶点的双靶点微管蛋白抑制剂已引起了许多研究者的兴趣,除上述微管蛋白双靶点外,还有微管蛋白与Hsp90、Src、PI3K、TDO/IDO和拓扑异构酶等靶点的双靶点抑制剂6。这些双靶点微管蛋白抑制剂通过调节微管蛋白动力学和另一个协同靶点的活性,就足以达到良好的治疗效果,更准确地靶向肿瘤组织,降低对正常组织的毒性,增强对肿瘤组织的治疗效果。关于抑制剂设计方面,除了传统的药物发现策略,不断的有许多新的方法被用于双靶点抑制剂的合理设计,例如,通过预测微管蛋白结合口袋与其他抗肿瘤靶点之间的结构相似性或通过机器/深度学习进行信号网络分析、计算机药效团建模、人工智能技术等。它们可用于识别具有新型骨架的双靶点先导结构,也可用于双靶分子的结构修饰,这些大大的推进了抗肿瘤药物的进展。
*药物信息来源于药渡数据库
参考文献:
1.Verrills, N. M.; Kavallaris, M. Improving the targeting of tubulin-binding agents: lessons from drug resistance studies. Curr. Pharm. Design 2005, 11, 1719-33.
2.Kavallaris, M. Microtubules and resistance to tubulin-binding agents. Nature reviews. Cancer 2010, 10, 194-204.
3.Canta, A.; Chiorazzi, A.; Cavaletti, G. Tubulin: a target for antineoplastic drugs into the cancer cells but also in the peripheral nervous system. Current medicinal chemistry 2009, 16, 1315-24.
4.Sun, D.; Zhao, Y.; Zhang, S.; Zhang, L.; Liu, B.; Ouyang, L. Dual-target kinase drug design: Current strategies and future directions in cancer therapy. Eur. J. Med. Chem. 2020, 188, 112025.
5.Poornima, P.; Kumar, J. D.; Zhao, Q.; Blunder, M.; Efferth, T. Network pharmacology of cancer: From understanding of complex interactomes to the design of multi-target specific therapeutics from nature. Pharmacol. Res. 2016, 111, 290-302.
6.Shuai, W.; Wang, G.; Zhang, Y.; Bu, F.; Zhang, S.; Miller, D. D.; Li, W.; Ouyang, L.; Wang, Y. Recen
*声明:本文由入驻新浪医药新闻作者撰写,观点仅代表作者本人,不代表新浪医药新闻立场。
